Charcot-Marie-Tooth Disease Type 1B (CMT1B) Guide
Understanding CMT1B
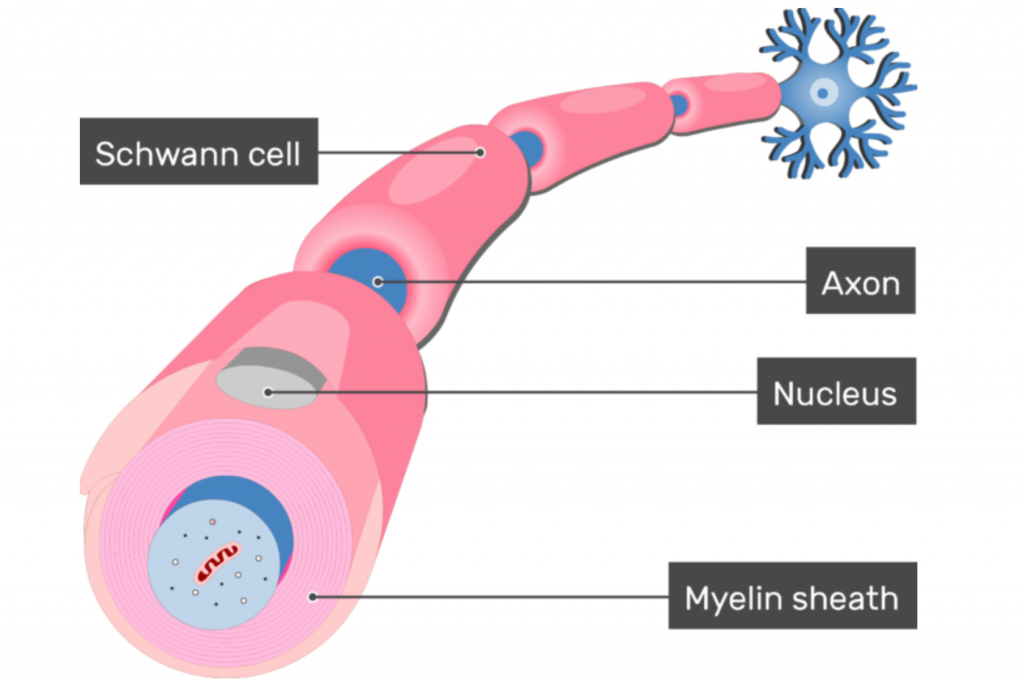
Charcot-Marie-Tooth disease type 1B (CMT1B) is a form of CMT that is inherited with autosomal dominance. This means affected individuals have at least one copy of the disease-causing gene and usually one normal copy of the gene on a pair of chromosomes that do not affect gender. In CMT1B, the part of the nervous system that is dysfunctional is called myelin. Created by cells called Schwann cells, myelin is a wrapping around the parts of the nerves that facilitate rapid electrical signals to other parts of the body. The myelin sheath is critical for the fast signaling our nerves are accustomed to, and the body’s entire communication system will break down without it. In CMT1B, that myelin sheath is dysfunctional because the Schwann cells have mutations within a gene called myelin protein zero, or MPZ — a necessary component of the myelin sheath.
CMT1B is the most common form of CMT caused by mutations in MPZ, however, there are other CMT subtypes also associated with MPZ mutations, including CMT2I, CMT2J and some forms of CMT3. MPZ mutations account for approximately 6% of American CMT patients.
Causes & Symptoms of CMT1B
The protein produced by the MPZ gene is exclusively found in Schwann cells — the cells that produce the myelin sheath in the peripheral nervous system. New evidence indicates that the abnormalities within the MPZ protein seen in some CMT1B patients can trigger the Schwann cell’s normal process of sequestering misfolded, and therefore dysfunctional, protein. This process of sequestering misfolded protein is called the “unfolded protein response” or the UPR. In healthy cells, this is exactly what should happen when there is a dysfunctional protein floating around that could cause problems. Unfortunately, in individuals with CMT1B who have abnormalities or mutations in their MPZ protein, this response is constantly working due to the abundant and continued production of their abnormal MPZ proteins. Over time, the UPR response in the Schwann cells become overwhelmed, resulting in sick, and ultimately dying, Schwann cells. An overworked UPR is not the cause of all CMT1B cases, but it is one known mechanism that can be targeted. Regardless, all CMT1B cases eventually result in sick and dying Schwann cells.
When a Schwann cell is sick, it cannot effectively produce the myelin sheath insulation to provide axonal protection that nerves require. When nerves start to lose bits and pieces of their myelin sheath (demyelination), the electrical communication constantly passing through the nerves begins to slow down. Slow signals are not inherently bad, but our muscles and our nervous system are accustomed to nerve signals being sent at a particular speed. If that speed is not matched, the system falls out of synchronization and signals eventually fail to reach their targets altogether. When this begins to happen, muscles will waste away. It is this wasting away, or atrophy, that results in disease symptoms, worsening over time and progressing up along an individual’s arms and legs. The muscle atrophy is one symptom of CMT1B, but this same weakening process simultaneously affects the individual’s sensory nerves as well. In the same regions of the body experiencing atrophy, CMT1B patients will likely also experience decreased sensation.
Read more about CMT signs and symptoms
Treatments for CMT1
For at least some CMT1B patients, the UPR is a likely target for treatments. A drug that can support the UPR and better facilitate its function in a chronic stress environment — like in the Schwann cells of CMT1B patients — has already been identified. The CMT Research Foundation is currently investing in research designed to further develop this drug, which has already demonstrated promising effects in animal models.
Bai, Y., Wu, X., Brennan, K. M., Wang, D. S., D’Antonio, M., Moran, J., … Shy, M. E. (2018). Myelin protein zero mutations and the unfolded protein response in Charcot Marie Tooth disease type 1B. Annals of Clinical and Translational Neurology, 5(4), 445–455. https://doi.org/10.1002/acn3.543
Morena, J., Gupta, A., & Hoyle, J. C. (2019). Charcot-Marie-Tooth: From Molecules to Therapy. International Journal of Molecular Sciences, 20(14). https://doi.org/10.3390/ijms20143419
Saporta, A. S. D., Sottile, S. L., Miller, L. J., Feely, S. M. E., Siskind, C. E., & Shy, M. E. (2011). Charcot-marie-tooth disease subtypes and genetic testing strategies. Annals of Neurology, 69(1), 22–33. https://doi.org/10.1002/ana.22166
Shy, M. E., Jáni, A., Krajewski, K., Grandis, M., Lewis, R. A., Li, J., … Kamholz, J. (2004). Phenotypic clustering in MPZ mutations. Brain : A Journal of Neurology, 127(Pt 2), 371–384. https://doi.org/10.1093/brain/awh048
Shy, M. E. (2006). Peripheral neuropathies caused by mutations in the myelin protein zero. Journal of the Neurological Sciences, 242(1-2 SPEC. ISS.), 55–66. https://doi.org/10.1016/j.jns.2005.11.015
Address
4062 Peachtree Road
Suite A209
Atlanta, GA 30319
Phone Number
404.806.7180
