Charcot-Marie-Tooth Disease Type 1 Guide
Understanding Charcot-Marie Tooth Disease Type 1
Charcot-Marie-Tooth Type 1 is just one of many different types of Charcot-Marie-Tooth disease. While the different CMT subtypes share similar symptoms, the underlying cause of each type of CMT varies. Understanding these causes is critically important for developing and administering treatments.
CMT1 is a form of CMT that is inherited with autosomal dominance (its inheritance pattern). This means the disease occurs with at least one copy of the disease-causing gene, and affected individuals usually also have one normal copy of the gene on a pair of chromosomes that do not affect gender. In CMT1, the part of the nervous system that is dysfunctional is called myelin. Myelin is a wrapping around the parts of the nerves that facilitate rapid electrical signals to other parts of the body. When it is damaged, like in CMT1, that signaling can slow, weaken and become uncoordinated. The slowing of electrical signals and loss of coordinated signaling is what leads to muscle weakness.
Symptoms of CMT1
The first symptom of CMT1 is usually muscle weakness in the feet. This presents as hammertoes, pes cavus (high arch) and other foot deformities, which are caused by the small muscles in the feet deteriorating. Additionally, people with CMT1 often have a loss of sensation accompanying this weakness. Their symptoms affect both motor and sensory function because the myelin that is meant to insulate nerves is damaged, impacting communication with the musculature and the sensory organs in the skin. Charcot-Marie-Tooth Type 1 is a progressive disorder, meaning the symptoms will worsen over time, but this progression is usually slow. Eventually, the weakness and loss of sensation will work its way closer to the torso and begin affecting the arms and hands. As the disease worsens, people with CMT1 can expect a loss of coordination or balance due to a loss of a particular kind of sensory ability called proprioception. Proprioception is the ability to sense where your body (and limbs) are in space. Loss of proprioception causes uncoordinated movements and imbalance.
Read more about CMT signs and symptoms
Causes of CMT1
Axons are what carry signals to the nerves; the nerves that move your toes or fingers reach all the way from your spinal cord to your fingers and toes. Axons are covered by an insulating material called myelin. Myelin is produced by cells called Schwann Cells, and the myelin wraps around the axon.
Type 1 Charcot-Marie-Tooth has been linked to several different genetic causes. Most of these are specific to Schwann Cells that produce myelin in the peripheral nervous system. The myelin produced by these cells is essential for normal function, therefore disruptions in the Schwann Cells and their ability to produce myelin frequently result in CMT1 disease symptoms. Because the defects caused by these mutations are unique to these specific cells, improving myelin function a strong target for designing therapies.
Need an even simpler way to understand CMT genetics? Watch this quick 5-minute video from scientific expert Dr. Grace Pavlath.
When genetic links are discovered, they are added to the CMT type name as an uppercase letter as a genetic identifier. For example, CMT1A, the most common form of CMT. There are seven different genetic types of CMT1:
- CMT1A: Duplication of the PMP22 gene
- CMT1B: Mutations within the MPZ gene
- CMT1C: Mutations within the gene known as either LITAF or SIMPLE
- CMT1D: Mutations within the EGR2 gene
- CMT1E: Mutations within the PMP22 gene
- CMT1F: Mutations within the NEFL gene
- CMT1G: Mutations within the PMP2 gene
- HNPP: Deletion of the PMP22 gene
As more genes are identified, the types will expand. And soon, scientists and researchers will primarily use the gene mutation to identify the problem rather than the subtype.

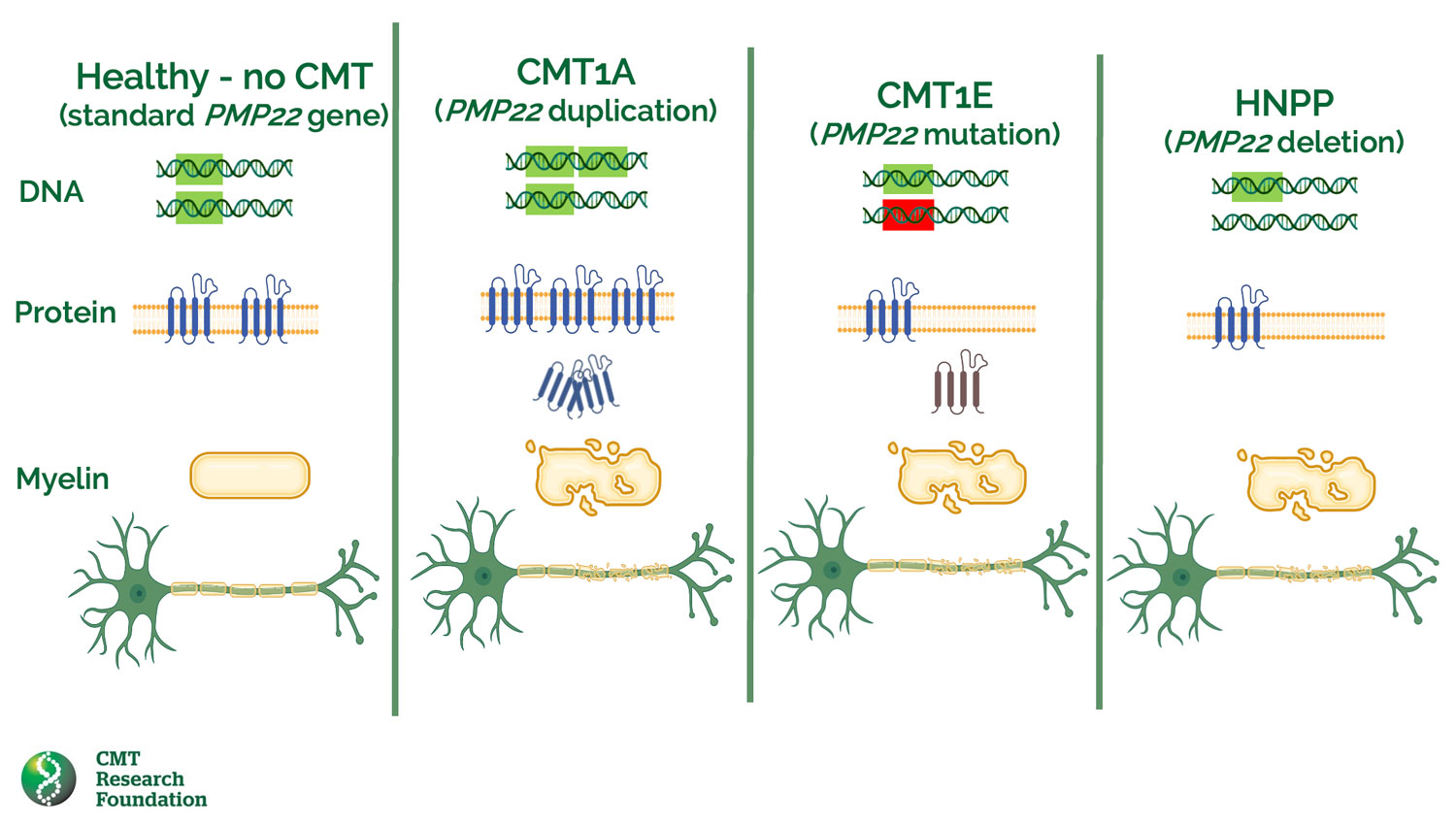
Do I have CMT1A, CMT1E or HNPP?
Among the different subtypes of CMT, CMT1A, CMT1E and HNPP (Hereditary Neuropathy with Pressure Predisposition) are linked to a single PMP22 gene, which is involved in the production of the PMP22 protein of the same name. However, they are distinguished by the type of mutation on PMP22 and the consequences of the disease.
- Caused by a duplication of the PMP22 gene.
- Duplication of the PMP22 gene leads to an overproduction of the PMP22 protein, which is essential for the structure of peripheral nerves.
- Characterized by demyelination of peripheral nerves, which means that the myelin sheath that insulates nerve fibers is damaged, affecting nerve conduction.
- Patients with CMT1A may experience symptoms of muscle weakness, muscle atrophy, and sensory neuropathy-related disturbances.
- Caused by mutations in the PMP22 gene.
- Unlike CMT1A, CMT1E is usually caused by point mutations or unduplicated mutations in the PMP22 gene.
- CMT1E shares some clinical features with CMT1A, including peripheral nerve demyelination and motor and sensory neuropathy-related symptoms.
- The mechanisms of CMT1E are not yet fully understood, but in general, patients with CMT1E have an earlier and more severe form of CMT.
HNPP (Hereditary Neuropathy with Liability to Pressure Palsies):
- Caused by a deletion (loss) of one copy of the PMP22 gene, instead of a duplication.
- This deletion leads to a reduction in the amount of PMP22 protein, which can make peripheral nerves more sensitive to pressure.
- Symptoms of HNPP often include muscle weakness and neuropathic pain that occurs in response to pressure or exertion, but these symptoms can be intermittent.
It is important to note that Charcot-Marie-Tooth disease is a group of complex genetic disorders, and there are many other subtypes of CMT, each resulting from mutations in different genes. The symptoms and severity of the disease can vary greatly from person to person, even within the same subtype of CMT. An accurate diagnosis and medical evaluation are essential to understanding the specific nature of the disease in a given person.
Treatments for CMT1
While no treatments or cures currently exist for CMT, the science to change that does. The CMT Research Foundation is currently funding cutting-edge research solely focused on drug development, including projects to advance treatments for Charcot-Marie-Tooth disease Type 1.
