Charcot-Marie-Tooth Disease Type 1E: A Guide
Understanding CMT1E
Charcot-Marie-Tooth disease type 1E is a rare type of CMT caused by point mutations in the pmp22 gene.
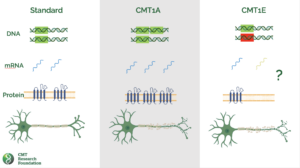
This gene encodes peripheral myelin protein 22 (PMP22), a major protein component of myelin, the protective sheath that insulates and supports nerve fibers.
Mutations in the PMP22 gene can lead to a variety of pathological changes in the peripheral nerves, including:
- Demyelination: This is the loss of myelin, which exposes and damages the underlying nerve axons. Demyelination can be caused by a variety of factors, including direct damage to myelin-producing cells (Schwann cells), or by abnormal accumulation of PMP22 protein in Schwann cells.
- Axonopathy: This damages the nerve axons themselves. Axonopathy can be caused by demyelination, or by other factors, such as abnormal protein accumulation in axons or impaired axonal transport.
- Segmental demyelination and remyelination: This is a characteristic feature of CMT1E, in which segments of nerve fibers are demyelinated and then remyelinated. This process can lead to the formation of “onion bulb” formations, which are concentric layers of myelin around axons.
The severity of the pathological changes in CMT1E can vary depending on the specific mutation in the PMP22 gene. In some cases, the changes are relatively mild and result in slowly progressive neuropathy. In other cases, the changes are more severe and result in a more rapidly progressive neuropathy with early onset.
The pathological changes in CMT1E lead to a number of clinical symptoms, including muscle weakness, atrophy, and sensory loss in the distal extremities (feet and hands). In some cases, there may also be involvement of the proximal extremities (arms and legs) and the cranial nerves.
There is no cure for CMT1E, but there are treatments that can help to manage the symptoms and improve quality of life. These treatments include physical therapy, occupational therapy, and orthopedic devices to support the feet and ankles. In some cases, pain medications or surgery may be necessary to treat complications of the disease.
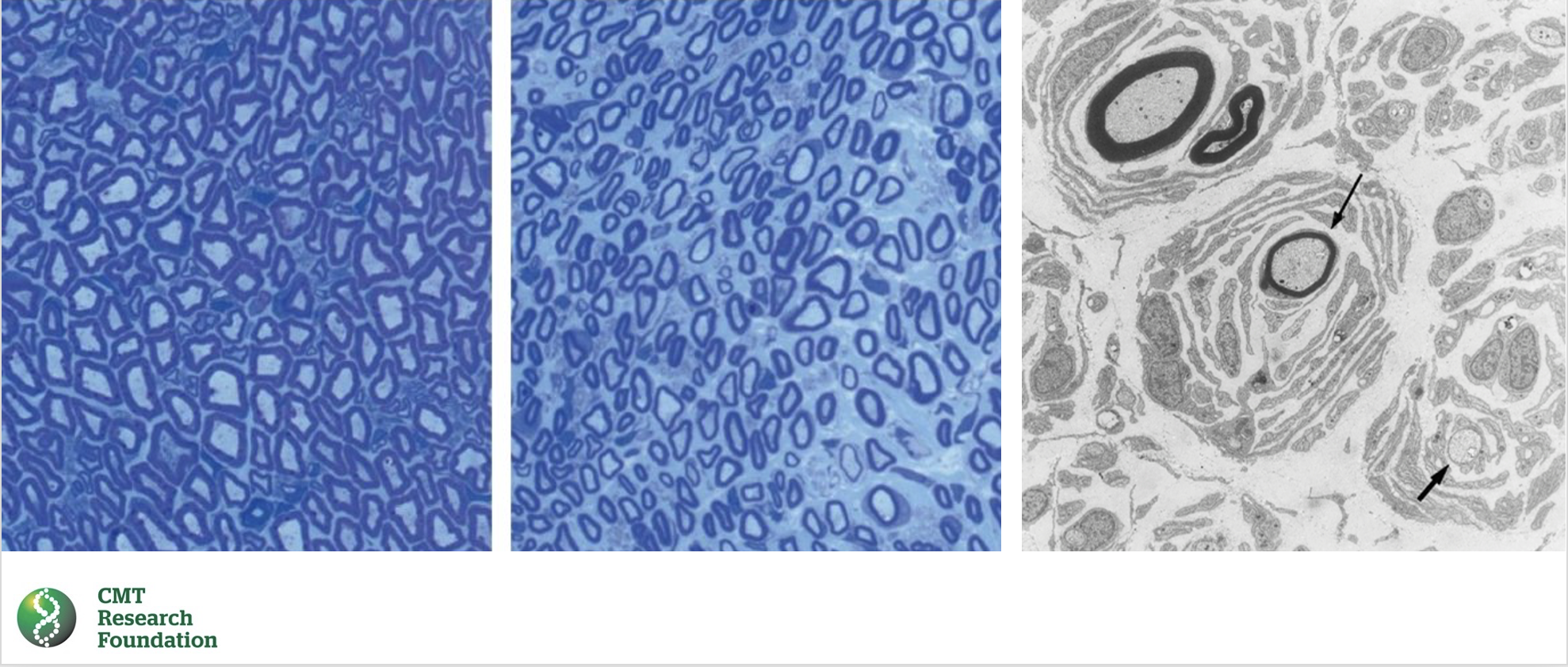
This illustration shows a normal peripheral nerve fiber on the left and a peripheral nerve fiber affected by CMT1E in the middle. The nerve fiber affected by CMT1E is demyelinated, meaning that the myelin sheath that insulates and protects the nerve fiber has been lost. This can be seen by the pale staining of the nerve fiber on the right. The myelin sheath is normally stained dark blue.
The illustration on the right shows onion bulb formations, which are concentric layers of myelin around axons. These formations are characteristic of CMT1E. The demyelination and onion bulb formations in CMT1E nerve pathology lead to damage of the nerve fibers, which causes the symptoms of the disease.
Source: NINDS: https://www.ninds.nih.gov/health-information/disorders/charcot-marie-tooth-disease
Treatments for CMT1E
There are currently no FDA-approved treatments for CMT1E. However, the development of drugs to treat other forms of CMT caused by pmp22 genetic defects are active areas of research interest. The CMT Research Foundation is pursing multiple projects that may have direct relevance to CMT1E, including attacking autophagy, delivering genetic therapies to the Schwann Cells, and addressing motor neuron deterioration.
Citations for the pathology of CMT1E:
- Vallat, J.-M., & Weiss, J. (2017). Peripheral Nerve Disorders: Pathology and Genetics. Springer.
- Dyck, P. J., & Thomas, P. K. (2005). Peripheral Neuropathy. Elsevier Health Sciences.
- Rossor, M. N., Polke, J. M., & Griffin, J. W. (2009). Bailey & Love’s Short Practice of Surgery. CRC Press.
- Berciano, J., Gallardo, E., & García-Arumí, E. (2016). Charcot-Marie-Tooth disease type 1E: Clinical and genetic characteristics. Revista de Neurología, 63(10), 449-457.
- Szigeti, K., & Lupski, J. R. (2009). Charcot-Marie-Tooth disease. Orphanet Journal of Rare Diseases, 4(1), 1-15.
One Woman’s Story
In 2006, I was diagnosed with CMT. Back then, I was playing field hockey and living a normal life. An athletic trainer who had taught my family called my mom, said something was off, and after genetic testing, we learned she was very very right. It wasn’t for many years later that I would get the 1E diagnosis. 1E is not well-known by clinicians; we’re often diagnosed as 1A. Once I started going to an INC doctor though, I was able to really be placed. Hearing I was rare among a rare disease wasn’t what I wanted, but here we are. If you have CMT 1E, we want you to join our small but fierce group of individuals working tirelessly for a cure. We’re going to do it. Join us. Read more.